Метилирование ДНК
Что такое метилирование ДНК – эпигенетическая модификация, регулирующая экспрессию генов
Знаете ли вы, что количество объятий, которые ребенок получает в детстве, может повлиять на профиль метилирования ДНК ребенка и на процесс развития?(1) Фактически, метилирование ДНК влияет на различные аспекты повседневной жизни, о которых большинство людей даже не подозревает, например, на процесс созревания плодов сладкого апельсина (2). Итак, что такое метилирование ДНК?
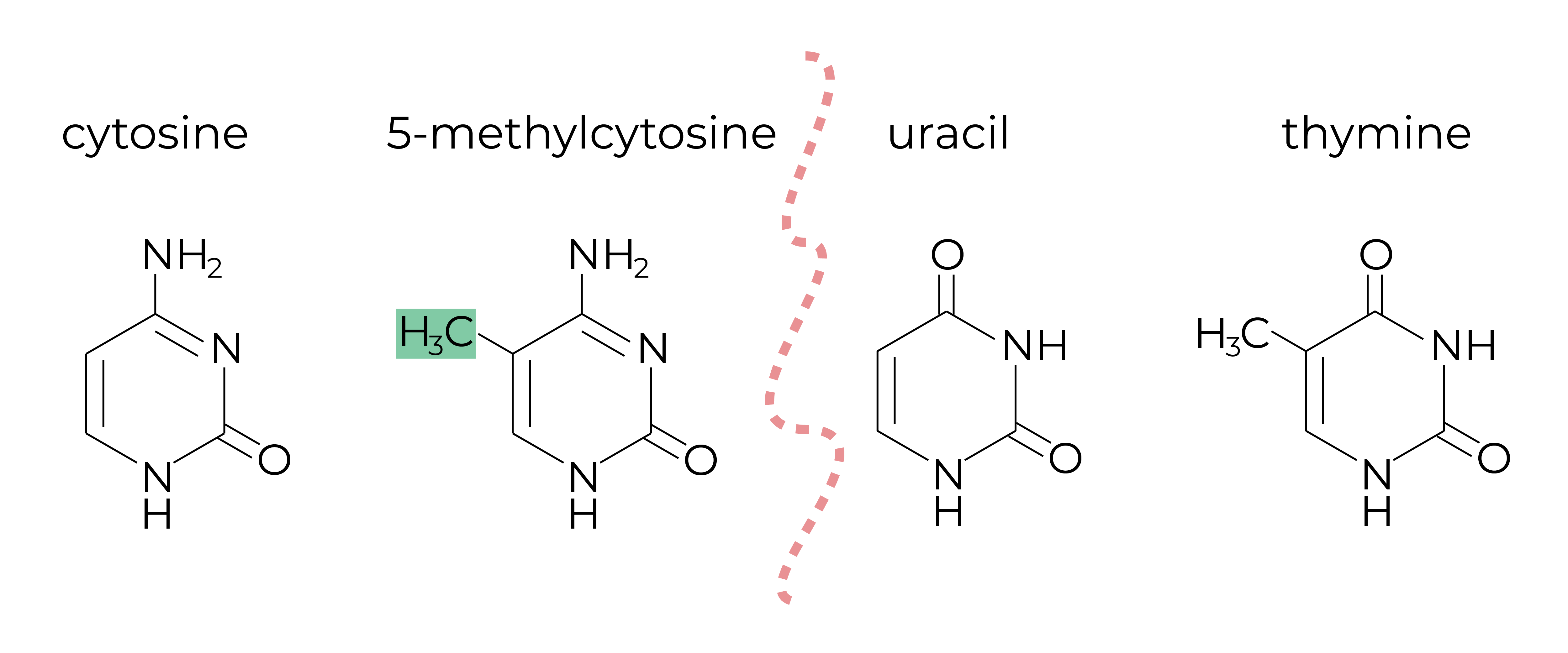
Метилирование ДНК - это химическая модификация молекул ДНК. В большинстве случаев метилирование ДНК заключается в ковалентном присоединении метильной группы к 5-С положению цитозина с образованием 5-метилцитозина (5mC) (рис. 1, две структуры слева). Эту реакцию осуществляют ферменты ДНК-метилтрансферазы, которые образуют и поддерживают паттерны метилирования ДНК в геноме. Метилирование ДНК регулирует экспрессию генов без изменения последовательности нуклеотидов. Следовательно, метилирование ДНК является эпигенетическим маркером, где «epi» означает, что эта особенность находится «на вершине» генетики, и выполняет свои задачи без изменений в последовательности ДНК.
Метилирование ДНК - один из наиболее широко изученных эпигенетических маркеров.(3) У эукариот метилирование ДНК повсеместно происходит в динуклеотидах CpG. Паттерны метилирования ДНК в промоторах, телах генов и энхансерах играют жизненно важную роль в регуляции экспрессии генов.(4-6)
Общепризнанные паттерны метилирования ДНК
Были широко изучены некоторые паттерны метилирования ДНК и собран достаточный объем знаний об их влиянии на экспрессию генов. Вот несколько таких примеров.
- Гиперметилирование ДНК (существенно повышенный уровень метилирования ДНК) в промоторных областях обычно связано с супрессией или молчанием генов. Хорошим примером является то, что гиперметилирование ДНК необходимо для инактивации Х-хромосомы и «молчанием» повторяющихся участков ДНК.(4)
- Сайты CpG на островках CpG (CGIs, регионы с высокой частотой сайтов GpG), расположенные в промоторах генов «домашнего хозяйства» и регуляторных генах, обычно устойчивы к метилированию ДНК. Однако было обнаружено, что эти промоторные CG-островки аберрантно гиперметилированы в генах-супрессорах опухолей и генах репарации ДНК, которые, в свою очередь, подавляют активацию и экспрессию соответствующих генов. Такой паттерн аберрантного метилирования ДНК в промоторных островках CGI является наиболее широко изученным эпигенетическим изменением при онкологии.(5,6)
- Было обнаружено, что CpG в энхансерах содержат паттерны метилирования ДНК, специфичные для тканей или клеток. Такие дифференциально метилированные области (DMR) могут способствовать уникальной экспрессии генов среди различных типов тканей и клеток.(7)
Учитывая, что точная регуляция генов важна для многих фундаментальных биологических процессов, метилирование ДНК необходимо на протяжении всего развития организма и гомеостаза. Таким образом, выяснение паттернов метилирования ДНК в образцах различных видов в различных условиях позволит проводить дальнейшее изучение его роли в механизмах регуляции генов. Кроме того, это расширит наше понимание того, как метилирование ДНК влияет на болезни и старение человека, а также на здоровье экосистем. Как же тогда нам определить профиль метилированной ДНК?
Как определить профиль метилированной ДНК
Золотым стандартом для анализа метилирования ДНК является бисульфитная конверсия. Бисульфитные реагенты превращают неметилированные остатки цитозина в урацилы (рис. 1, третья формула), оставляя метилированные остатки в исходном состоянии. Во время ПЦР ДНК-полимераза распознает урацилы как тимины, и последующий анализ определяет «C» как метилированные цитозины, а «T» (рис. 1, последняя формула) как неметилированные цитозины в образце ДНК. На основе бисульфитной конверсии было разработано множество методов для исследования метилирования ДНК. Поскольку возросла потребность в полногеномном секвенировании метилированной ДНК, методы, основанные на NGS, стали очень популярными.
Методы, основанные на NGS, являются наиболее исчерпывающими и недавно стали основным средством анализа метилирования ДНК. Методы на основе NGS дают результаты с высоким охватом по всему геному и совместимы с любыми видами организмов. Таким образом, бисульфитное секвенирование приобретает огромную популярность среди исследователей для определения профиля метилированной ДНК, о чем свидетельствует рост числа соответствующих публикаций за последние годы. Образцы ДНК, конвертированные бисульфитом, необходимо конструировать в библиотеки, содержащие адаптеры для секвенирования на совместимом приборе NGS. Метилированные цитозины определяются как «C» в результате проведения NGS.
К настоящему времени уже реализовано множество методов бисульфитного секвенирования, которые позволяют получить информацию о метилировании ДНК, необходимую для различных аспектов в биологии. Наиболее широко используемые методы NGS включают бисульфитное секвенирование ограниченного набора геномных локусов (RRBS), полногеномное бисульфитное секвенирование (WGBS) и таргетное бисульфитное секвенирование. Эти методы имеют общие этапы в процедуре подготовки библиотеки, такие как бисульфитная конверсия, лигирование адаптеров и ПЦР-амплификация, но немного отличаются в начале методики подготовки исходного материала ДНК.
Бисульфитное секвенирование ограниченного набора геномных локусов
Процедура RRBS основана на использовании рестриктазы для обогащения пула ДНК фрагментами с участками CpG всего генома перед проведением стандартной процедуры бисульфитной конверсии и создания библиотеки.
Особенности RRBS:
- Процедура уменьшает число необходимых раундов секвенирования для получения аналогичных по качеству данных с методами полногеномного бисульфитного секвенирования (WGBS), позволяет выявлять метилированные участки ДНК наиболее важных регуляторных областей, таких как промоторы, CpG-островки и тел генов. Приблизительно 10-20% ридов, которые обычно требуются для проведения WGBS, в методе RRBS могут покрывать ≥70% промоторов, CpG-островков, тел генов и около 35% энхансеров.
- RRBS является самым экономичным методом по сравнению с WGBS и будет полезен для полногеномного скрининга метилирования ДНК в крупномасштабных исследованиях
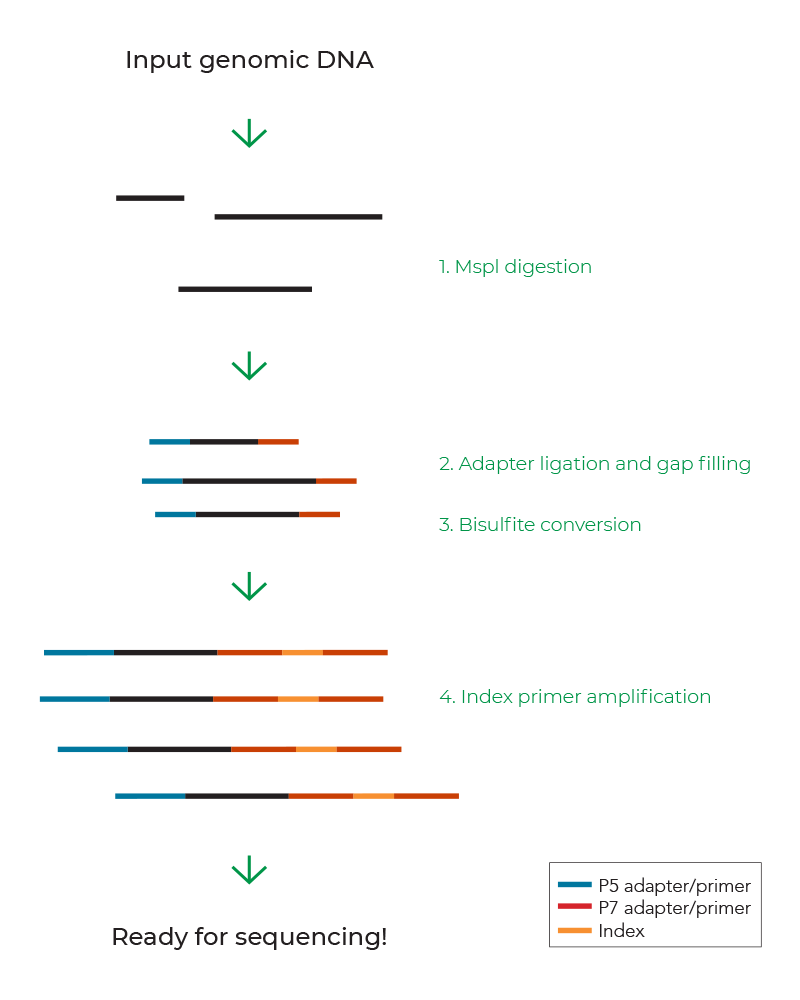
За прошедшие годы появилось множество вариантов RRBS, пример базового рабочего процесса RRBS на рисунке 2 описывает основные этапы подготовки библиотек RRBS из исходной геномной ДНК: расщепление MspI, лигирование адаптеров, бисульфитная конверсия и ПЦР-амплификация с индексированными праймерами. Специальный набор имеет простой протокол и требует всего 10 нг геномной ДНК, что делает метод RRBS более доступным для всех пользователей.
Полногеномное бисульфитное секвенирование
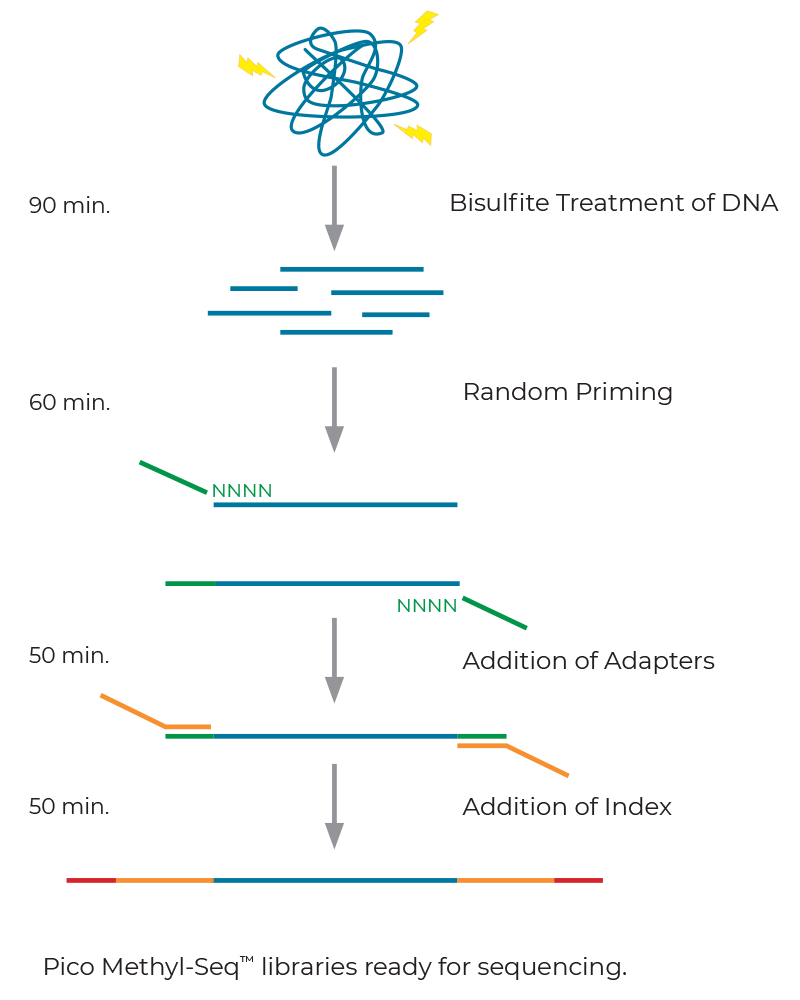
Доступно множество протоколов для проведения полногеномного бисульфитного секвенированя (WGBS). В основном, протокол включает этапы фрагментации геномной ДНК, бисульфитной обработки, лигирования адапторов и амплификации с индекс-праймерами.
Особенности WGBS
- WGBS определяет статус метилирования всех цитозинов исходного образца, давая информацию о полном профиле метилированной ДНК всего генома. Ученые, используя данные WGBS, с большей вероятностью получат de novo информацию о новых паттернах метилирования ДНК.
- В зависимости от размера генома, для получения значительной глубины и точности метода WGBS необходимо получить несколько сотен миллионов ридов. Поэтому стоимость секвенирования оказывается значительно большей.
- Основа метода WGBS показана на рис.3.Набор Pico Methyl-SeqLibrary Prep Kit особенно подходит для небольшого количества геномной ДНК, от 10 пг.
Таргентное бисульфитное секвенирование
Помимо двух ранее упомянутых методов профилирования метилированной ДНК всего генома, таргетное бисульфитное секвенирование позволяет обнаруживать сайт-специфическое метилирование ДНК в желаемых локусах.
Особенности таргетного бисульфитного секвенирования
- Самый прямой способ - разработка NGS-совместимых ПЦР-праймеров для определенных областей гена, представляющих интерес. Это позволяет исследовать уровни метилирования ДНК только в выбранных областях генома, что снижает затраты на секвенирование образца по сравнению с полногеномными методами.
- Таргетное бисульфитное секвенирование - отличный вариант для проверки данных, полученных с помощью других полногеномных методов, а также для скрининга множественных участков генов в большой выборке. С помощью этого метода ученые могут исследовать от 1 до 192 регионов и более в одном образце.
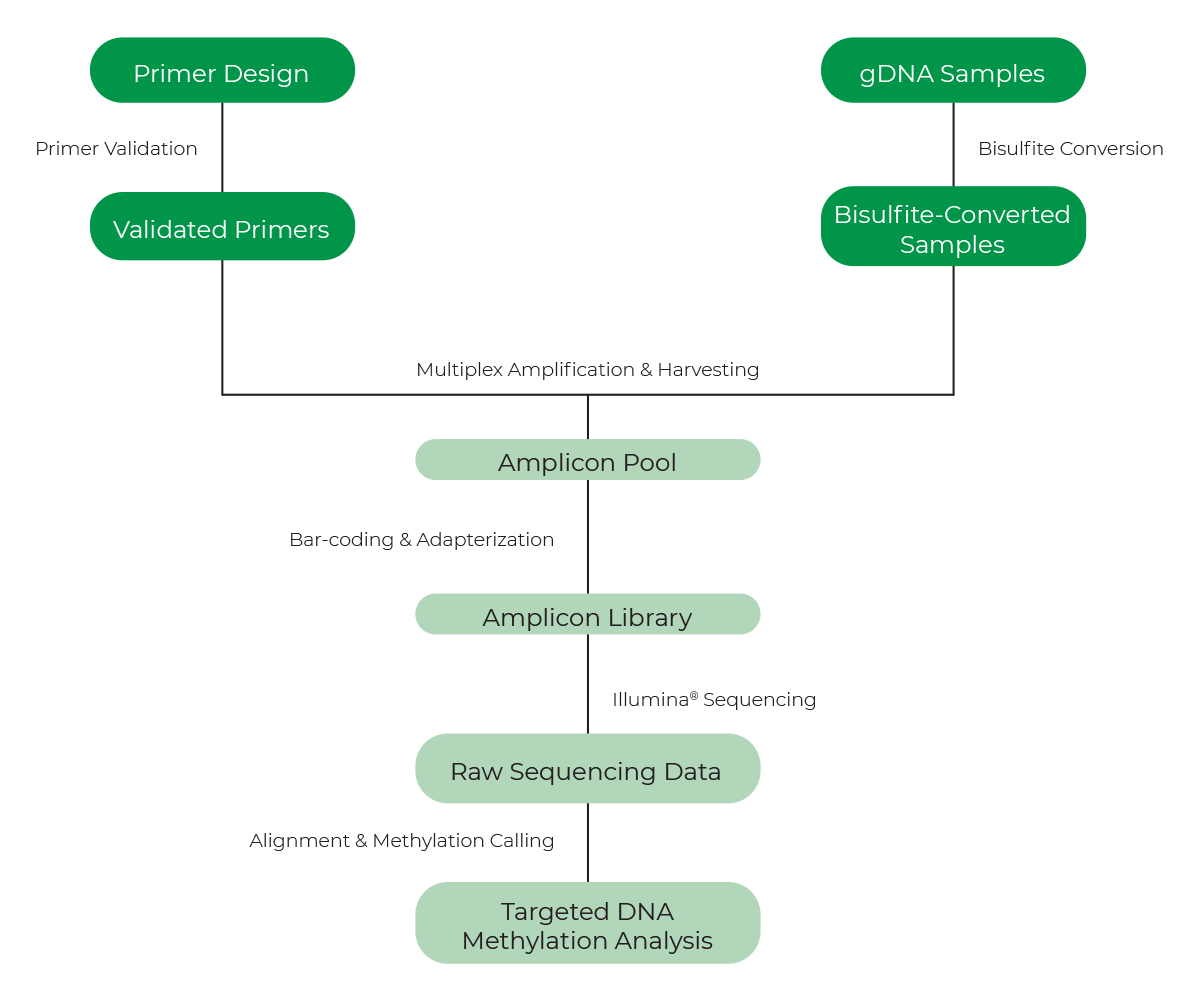
Общий протокол таргетного бисульфитного секвенирования, доступный от ZymoResearch, показан на рисунке 4.
Изучение метилирования ДНК с помощью NGS является мощным инструментом
Исследование метилирования ДНК с помощью NGS оказалось очень плодотворным. Ученые постоянно совершенствуют методы NGS, а также связанные с ними биоинформационные данные всех типов бисульфитного секвенирования, чтобы расширить сферу их применения.
- Исследователи применили WGBS для выявления гетерогенности метилирования генома одиночных клеток печени мыши (8). В среднем, было проанализировано 21,6 миллиона сайтов CpG в тотальном образце и 2,2 миллиона сайтов CpG в образце отдельных клеток.
- Ученые использовали RRBS для картирования метилирования ДНК в образцах курицы, опоссума и утконоса.(9) Эти данные впервые продемонстрировали участие метилирования ДНК в инактивации Х-хромосомы у сумчатых млекопитающих.
- Метод RRBS также использовался для обнаружения биомаркеров метилирования ДНК при состоянии пищевода Барретта. Этот синдром является важным фактором риска развития рака пищевода.(10) После того, как был определен биомаркерный участок CpG после проведения RRBS 46 образцов биопсии, таргетное бисульфитное секвенирование стали применять для проверки и расширения значимых сайтов CpG в свежих и архивные клинических образцах.
Методы с использованием микрочипов. До стремительного развития NGS стандартным методом профилирования метилированной ДНК было использование микрочипов. На этой платформе было получено множество данных. Кратко: ДНК, обработанную бисульфитом, денатурируют для гибридизации с CpG-специфическими зондами, иммобилизованными на матрице. Меченые дидезоксинуклеотиды включаются во время элонгации для получения неметилированных и метилированных сигналов, которые используются для определения уровня метилирования в каждом участке CpG.
Несмотря на то, что такие методы надежны в использовании и удобны для сравнения информации из разных наборов данных, они имеют несколько ограничений:
- Охват сайтов CpG в интересующем геноме в значительной степени ограничен. Массив MethylationEpic покрывает только ~ 3% от общего количества сайтов CpG в геноме человека.
- Низкая адаптивность в отношении видов образцов, поскольку один массив данных совместим только с одним конкретным видом животных.
В результате методы определения метилирования ДНК на основе NGS превзошли методы, основанные на использовании микрочипов и привлекают все большее внимание. Измерения метилирования ДНК с помощью NGS расширили наши знания о многогранной роли процесса метилирования ДНК. По мере того, как методологии продолжают совершенствоваться, возможности анализа метилирования ДНК с помощью NGS, несомненно, будут использоваться для создания более информативных данных, которые помогут собрать фрагменты головоломки в эпигенетике и за ее пределами.
В таблице представлена продукция и сервисы Zymo Research для изучения метилирования ДНК – комплексное решение для определения профиля метилированной ДНК.
|
Методики |
Рекомендуемые наборы и услуги |
|
Секвенирование ограниченного набора локусов (RRBS) |
|
|
Полногеномное бисульфитное секвенирование (WGBS) |
Pico Methyl-Seq Library Prep Kit |
|
Таргентное бисульфитное секвенирование |
|
|
Бисульфитная конверсия |
EZ DNA Methylation-Lightning Kit (доступны пробные наборы) |
|
Бисульфитная конверсия без выделения ДНК |
EZ DNA Methylation-Direct Kits (доступны пробные наборы) |
|
Выделение ДНК |
Quick-DNA Kits (доступны пробные наборы) |
|
Очистка ДНК |
Genomic DNA Clean & Concentrator (gDCC) (доступны пробные наборы) DNA Clean & Concentrator Kits (DCC) (доступны пробные наборы) |
|
DNA Standards for Methylation Study |
Литература:
Moore, S. R.; McEwen, L. M.; Quirt, J.; Morin, A.; Mah, S. M.; Barr, R. G.; Boyce, W. T.; Kobor, M. S., Epigenetic correlates of neonatal contact in humans. Dev Psychopathol 2017, 29 (5), 1517-1538.
Huang, H.; Liu, R.; Niu, Q.; Tang, K.; Zhang, B.; Zhang, H.; Chen, K.; Zhu, J. K.; Lang, Z., Global increase in DNA methylation during orange fruit development and ripening. Proc Natl Acad Sci U S A 2019, 116 (4), 1430-1436.
Smith, Z. D.; Meissner, A., DNA methylation: roles in mammalian development. Nat Rev Genet 2013, 14 (3), 204-20.
Greenberg, M. V. C.; Bourc'his, D., The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol 2019, 20 (10), 590-607.
Flavahan, W. A.; Gaskell, E.; Bernstein, B. E., Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357 (6348).
Ehrlich, M., DNA hypermethylation in disease: mechanisms and clinical relevance. Epigenetics 2019, 14 (12), 1141-1163.
Song, Y.; van den Berg, P. R.; Markoulaki, S.; Soldner, F.; Dall'Agnese, A.; Henninger, J. E.; Drotar, J.; Rosenau, N.; Cohen, M. A.; Young, R. A.; Semrau, S.; Stelzer, Y.; Jaenisch, R., Dynamic Enhancer DNA Methylation as Basis for Transcriptional and Cellular Heterogeneity of ESCs. Mol Cell 2019, 75 (5), 905-920.e6.
Gravina, S.; Dong, X.; Yu, B.; Vijg, J., Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biol 2016, 17 (1), 150.
Waters, S. A.; Livernois, A. M.; Patel, H.; O'Meally, D.; Craig, J. M.; Marshall Graves, J. A.; Suter, C. M.; Waters, P. D., Landscape of DNA Methylation on the Marsupial X. Mol Biol Evol 2018, 35 (2), 431-439.
Moinova, H. R.; LaFramboise, T.; Lutterbaugh, J. D.; Chandar, A. K.; Dumot, J.; Faulx, A.; Brock, W.; De la Cruz Cabrera, O.; Guda, K.; Barnholtz-Sloan, J. S.; Iyer, P. G.; Canto, M. I.; Wang, J. S.; Shaheen, N. J.; Thota, P. N.; Willis, J. E.; Chak, A.; Markowitz, S. D., Identifying DNA methylation biomarkers for non-endoscopic detection of Barrett's esophagus. Sci Transl Med 2018, 10 (424).

Похожие статьи
Эпигенетика
Эпигенетика это наука об
изменениях в экспрессии генов, вызванных механизмами, не затрагивающими
последовательность ДНК. Но что именно это означает?
Все о бисульфитной конверсии
Краткое описание и основные моменты методики
Рекомендуемые товары

D5001 Набор для бисульфитной конверсии EZ DNA Methylation Kit, 50 реакций, Zymo Research
Набор EZ DNA Methylation Kit ..
0 руб.

D5005 Набор для бисульфитной конверсии EZ DNA Methylation-Gold Kit, 50 реакций, Zymo Research
Набор EZ DNA Methylation-Gold ..
0 руб.

D5020 Набор для бисульфитной конверсии EZ DNA Methylation-Direct Kit, 50 реакций, Zymo Research
Набор EZ DNA Methylation-Direc..
0 руб.

D5030 Набор для бисульфитной конверсии ДНК короткий протокол EZ DNA Methlyation-Lightning Kit, 50 реакций, Zymo Research
Набор EZ DNA Methlyation-Light..
0 руб.

D5450 Набор для подготовки библиотек RRHP 5-hmC Library Prep Kit, 12 реакций, Zymo Research
Набор RRHP 5-hmC Library Prep ..
0 руб.
D5451 Набор для подготовки библиотек RRHP 5-hmC Library Prep Kit, 25 реакций, Zymo Research
Набор RRHP 5-hmC Library ..
0 руб.

D5455 Набор для создания библиотек Pico Methyl-Seq Library Prep Kit, 10 реакций, Zymo Research
Pico Methyl-Seq Library Prep K..
0 руб.
D5456 Набор для создания библиотек Pico Methyl-Seq Library Prep Kit, 25 реакций, Zymo Research
Pico Methyl-Seq Library Prep K..
0 руб.

D5460 Набор для создания библиотек для бисульфитного секвенирования Zymo-Seq RRBS Library Kit, 24 реакции, Zymo Research
Zymo-Seq RRBS Library Kit - эт..
0 руб.
 DNA Set, 5 мкг/20 мкл, Zymo Research")
D5013 Набор метилированной и неметилированной ДНК человека Human Methylated & Non-Methylated (WGA) DNA Set, 5 мкг/20 мкл, Zymo Research
Набор Human Methylated & N..
0 руб.

D5040 Набор для бисульфитной конверсии ДНК на магнитных частицах EZ-96 DNA Methylation MagPrep, 4 x 96 реакций, Zymo Research
Набор для бисульфитной конверс..
0 руб.

D5014 Набор метилированной/неметилированной ДНК человека Human Methylated & Non-methylated DNA Set, 5 мкг/20 мкл, Zymo Research
Набор Human Methylated & N..
0 руб.

D5011 Универсальный стандарт метилированной ДНК человека Universal Methylated Human DNA Standard, 5 мкг, Zymo Research
Universal Methylated Human DNA..
0 руб.
D5012 Универсальный стандарт метилированной ДНК мыши Universal Methylated Mouse DNA Standard, 5 мкг, Zymo Research
Universal Methylated Mouse DNA..
0 руб.
-80x80.jpg "D5015 Стандарт метилированной конвертированной ДНК человека Bisulfite-Converted Universal Methylated Human DNA Standard, 1 мкг/50 мкл, Zymo Research")
D5015 Стандарт метилированной конвертированной ДНК человека Bisulfite-Converted Universal Methylated Human DNA Standard, 1 мкг/50 мкл, Zymo Research
Bisulfite-Converted Universal ..
0 руб.

D5405 Набор стандартов ДНК 5-Methylcytosine & 5-Hydroxymethylcytosine DNA Standard Set, 1 набор, Zymo Research
Набор стандартов ДНК 5-Methylc..
0 руб.

D5016 Стандарт неметилированной ДНК E. coli Non-Methylated Genomic DNA, 5 мкг/20 мкл, Zymo Research
Стандарт неметилированной ДНК ..
0 руб.

D5017 Набор метилированной и неметилированной ДНК с праймерами Methylated & Non-methylated pUC19 DNA Set, 20 нг, Zymo Research
Набор метилированной и неметил..
0 руб.

D5465 Набор для синтеза библиотек для полногеномного бисульфитного секвенирования Zymo-Seq WGBS Library Kit, 24 реакции, Zymo Research
Набор Zymo-Seq WGBS Library Ki..
0 руб.
Теги: Метилирование ДНК, эпигенетика